지중해빈혈
이론과 하이라이트 히스토리를 확인 할 수 있어요.
: Thalassemia
Hemoglobin 합성에 유전적 결함이 생기는 질환으로, 동남아시아 혈통이 있는 환자에서 발견되는 microcytic anemia의 형태로 국시에 주로 출제된다. PB smear가 IDA와 유사하므로 감별진단에 있어 주의해야 하며, 혈색소 전기영동검사 결과를 읽을 줄도 알아야 한다.
참고: 빈혈의 분류, 소적혈구빈혈의 비교
1. 개요
1) 정의: 유전적 문제로 헤모글로빈을 구성하는 α, β globin의 수가 불충분해지는 질환군
2) 역학: 동남아시아, 지중해 지역, 중동, 아프리카 등지에서 많이 발견됨
* 우리나라의 경우 동남아시아 출신 이민자들과 혼혈인들이 많아짐에 따라 국시에서도 중요도가 커지고 있다. 국시에서는 동남아시아 출신, 태국인 등으로 증례에 주어진다.
3) 병태생리
(1) 정상 hemoglobin의 구조
① Hemoglobin의 globin은 tetramer(= 2 alpha chains + 2 non-alpha chains)
② Non-alpha chain 종류에 따라 HbA, HbA2, HbA1C, HbF로 분류됨
③ 태아 상태에서는 HbF의 비중이 높으나, 출생 후 γ 대신 β가 만들어지면서 HbA로 대체됨
Hb | 구조 | % |
A | α2β2 (α 2개, β 2개) | 92 |
A2 | α2δ2 (α 2개, δ 2개) | 2.5 |
A1C(당화혈색소) | α2(β-N-glucose) | 3 |
F(fetal) | α2γ2 (α 2개, γ 2개) | < 1 |
(2) α thalassemia: α globin 생산량 감소
① α thalassemia trait: 4개 gene 중 1~2개 mutation
② HbH disease: 4개 gene 중 3개 mutation → β globin 4개 모인 HbH(β4) 형성
③ Hydrops fetalis with Hb Barts: 4개 gene 모두 mutation → γ globin 4개 모인 Hb Barts(γ4) 형성
(3) β thalassemia: β globin 생산량 감소
① 분류
• β thalassemia major: 2 severe mutation → β globin 생성 0
- 매우 심한 빈혈 → 지속적 수혈치료 필요
• β thalassemia intermedia: 1 severe, 1 mild mutation
- Major보다는 경한 빈혈 → 항상 수혈치료가 필요하지는 않음
• β thalassemia minor: 1 mutation, 1 정상 allele (carrier)
- 특별한 증상 없이 PB smear 이상 정도만 발생
② β globin 양 부족
• RBC 내부 Hb↓(hypochromic), mature RBC 수↓
• 따라서 HbA↓ / HbA2↑, HbF↑
③ α globin 축적
• RBC 내부에 inclusion body 형태로 축적
• 따라서 RBC membrane이 불안정해지며 hemolysis
• 따라서 splenomegaly & indirect hyperbilirubinemia
④ 빈혈로 인해 골수가 조혈작용을 위해 자극됨 → BM expansion → 뼈가 비대해지고 약해짐
2. 임상양상
Mutation 개수에 따라 증상/징후의 정도가 달라진다. β thalassemia minor의 경우 별다른 증상 없이 CBC상 빈혈만 관찰되는 경우가 많지만, β thalassemia major나 HbH disease 등의 경우 평생 수혈치료가 필요한 정도의 빈혈이 동반되거나 태아 단계에서 사망할 수도 있다.
1) 빈혈 양상: 피로, 운동 시 호흡곤란, 창백
2) 용혈성 빈혈 양상
(1) 황달: Indirect hyperbilirubinemia로 인해 발생
(2) 간비비대(hepatosplenomegaly)
3) 골수 비대로 인한 뼈의 변형: Chipmunk face(좌), hair-on-end skull(우)
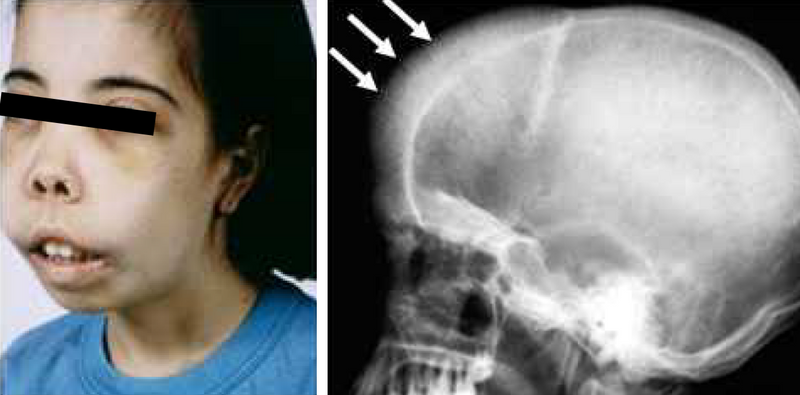
3. 검사소견
1) CBC: Microcytic hypochromic anemia
(1) Hb↓, Hct ↓
(2) RBC index: MCV↓, MCH↓, RDW 수치는 상이
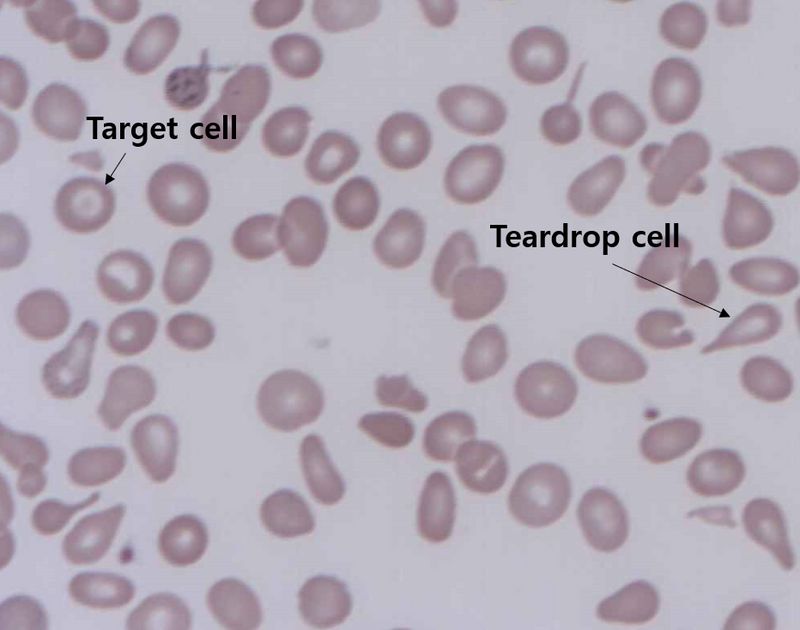
2) PB smear
(1) IDA와 유사한 microcytic hypochromic RBC
(2) Target cell, teardrop cell 등
4. 진단
1) 혈색소전기이동(Hb electrophoresis)
(1) 원리: Hb type에 따라 분자량이 달라 전기영동을 통해 A, A2, F 등으로 구분 가능
(2) α thalassemia
① α thalassemia trait: 정상
② HbH disease, hydrops fetalis with Hb Barts: 각각 HbH↑, Hb Barts↑
(3) β Thalassemia
① β thalassemia trait: HbA↓, HbA2↑, HbF↑
② β thalassemia major: HbA 없음, HbA2↑↑, HbF↑↑
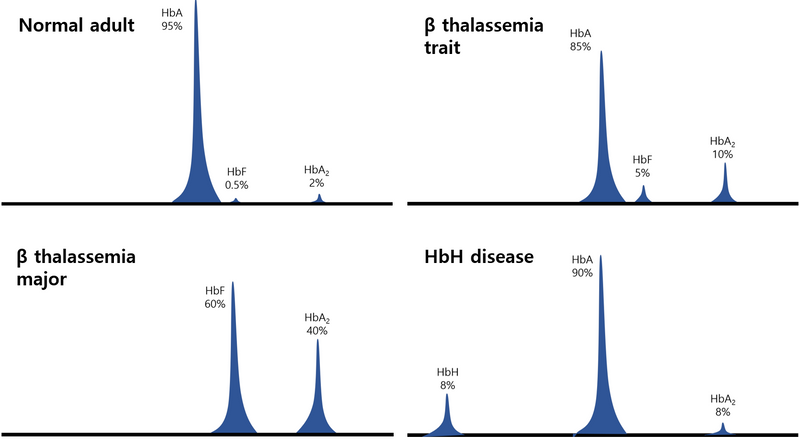
위 그림의 수치는 환자에 따라 달라질 수 있으니 대략적인 패턴 위주로 보아야 한다.
2) 유전자 검사
5. 치료
치료법은 적혈구 수혈과 조혈모세포이식 뿐이다.
1) 농축적혈구 수혈 + folic acid replacement
(1) 수혈 적응증: Hb < 7 g/dL
(2) 만성, 반복적 수혈로 인한 iron overload가 심해질 경우 iron chelation therapy 시행
• Deferoxamine(IV), deferasirox(PO) 등 투여
2) 조혈모세포이식
(1) Hypersplenism이 있을 때 splenectomy도 고려할 수 있지만, 궁극적인 치료를 위해서는 이식해야 함
(2) 간질환 없는 젊은 환자에서 적절한 공여자가 있다면 약 >80%의 완치율을 보임
Harrison 21e, pp.761-764